
Retrouvez tous vos contenus sur mobile avec l'application du Point Vétérinaire.
Téléchargez gratuitement l'application !


Retrouvez tous vos contenus sur mobile avec l'application du Point Vétérinaire.
Téléchargez gratuitement l'application !
Article de synthèse
Auteur(s) : Valérie Deniau*, Anne Couroucé-Malblanc**
Fonctions :
*Clinique vétérinaire de Grosbois, domaine
de Grosbois, 94470 Boissy-Saint-Léger
**Pôle équin, Oniris, École nationale
vétérinaire, agroalimentaire et de l’alimentation
de Nantes-Atlantique, Atlanpôle La Chantrerie
BP 44607, 44307 Nantes Cedex 3
Très fréquents chez les chevaux atteints d’une affection digestive inflammatoire ou opérés de coliques, les troubles de l’hémostase peuvent influer sur le pronostic vital de l’animal. Il convient donc de connaître les mécanismes de ce processus, ainsi que les principales maladies qui peuvent en compromettre le fonctionnement.
Les troubles de l’hémostase représentent chez le cheval, comme chez les autres espèces, une entité pathologique à la fois très diverse et incomplètement cernée. Ils peuvent être suspectés de façon très ponctuelle chez un animal en bonne santé présentant une tendance inhabituelle aux saignements, ou apparaître au cours de l’évolution d’une affection septique ou métabolique sévère. Des études à large échelle ont mis en évidence une prévalence très significative des déséquilibres de l’hémostase chez les chevaux atteints d’une affection digestive inflammatoire ou opérés de coliques, et souvent une relation avec le pronostic vital [6, 9]. Ces troubles sont également observés lors d’une affection septique ou de syndrome paranéoplasique [21]. Enfin, leur présence peut compromettre la réalisation ou les suites d’un acte chirurgical ou diagnostique invasif.
Pour autant, l’exploration de la fonction hémostatique n’est pas facilement menée en pratique. Les raisons en sont sans doute des indications moins fréquentes que celles des examens biochimiques de routine, une certaine complexité dans le choix et l’interprétation des analyses, un intérêt peut-être mal perçu et la disponibilité parfois restreinte de tests adaptés à l’espèce équine.
Dans cette première partie sont présentés les mécanismes de l’hémostase et de sa régulation, et les principales maladies qui peuvent en compromettre le fonctionnement chez le cheval.
L’hémostase physiologique désigne l’ensemble des mécanismes cellulaires et biochimiques qui assurent en permanence le contrôle des saignements consécutifs aux effractions vasculaires et le maintien de la fluidité sanguine dans tout le réseau circulatoire.
Trois étapes essentielles se succèdent (figure 1) :
– la formation rapide d’un amas dit “clou” plaquettaire : hémostase primaire ;
– puis sa consolidation par formation d’un réseau de fibrine et d’un caillot sanguin : hémostase secondaire ou coagulation sanguine ;
– enfin, un processus de fibrinolyse qui intervient par la suite, à la fois pour limiter l’extension du caillot et assurer son élimination progressive après réparation des parois vasculaires lésées.
L’hémostase primaire fait essentiellement intervenir les plaquettes sanguines ou thrombocytes.
Favorisée par un bref vasospasme avec ralentissement du flux sanguin, l’adhésion des plaquettes sur la brèche vasculaire est initiée par leur contact avec le collagène et les fibres élastiques sous-endothéliales. Elle est dans un premier temps réversible.
L’activation plaquettaire entraîne ensuite une agrégation définitive en présence de fibrinogène et de calcium, pour former le clou plaquettaire dont les constituants fusionnent (encadré 1).
La coagulation sanguine résulte de l’activation en cascade de multiples facteurs enzymatiques présents dans le plasma sous forme de précurseurs inactifs [7, 12, 17].
Elle peut être déclenchée par deux voies :
– la voie extrinsèque, dominante in vivo, est initiée lors de la libération du facteur III tissulaire ou thromboplastine par les cellules endothéliales et sous-endothéliales en regard de la lésion vasculaire ;
– la voie intrinsèque ne faisant intervenir que des éléments sanguins est déclenchée par le contact des facteurs de coagulation avec des surfaces de polarité négative comme les phospholipides des membranes plaquettaires et le collagène endothélial, ou avec les parois des tubes de prélèvement. In vivo, cette phase de contact est d’importance secondaire. Les déficiences en facteur XII entraînent rarement des troubles de l’hémostase cliniquement décelables. La voie intrinsèque est activée essentiellement par une rétroactivation de la thrombine sur les facteurs V, XI et VIII (figure 2) [26].
Les points clés de l’hémostase secondaire sont :
– une organisation en cascade qui permet une amplification maximale des réactions chimiques ;
– l’importance de certaines molécules “carrefour” comme le facteur X et la thrombine ;
– la nécessité d’ions calcium à de nombreuses étapes, qui explique les propriétés anticoagulantes des chélateurs du calcium (oxalates, citrate, EDTA) utilisés pour la conservation des échantillons sanguins.
L’ensemble des réactions aboutit à une voie commune de la cascade enzymatique de coagulation, qui s’achève par l’hydrolyse du fibrinogène en fibrine soluble puis insoluble, formant un caillot sanguin stable en regard du clou plaquettaire.
Ce caillot se rétracte ensuite en 20 à 60 minutes et assure l’étanchéité de la paroi vasculaire jusqu’à la réparation de cette dernière.
Dernière étape de l’hémostase physiologique, la fibrinolyse, dissolution de la fibrine et disparition du caillot, permet la restauration complète de la perméabilité vasculaire. Son agent essentiel est la plasmine, enzyme dérivée de l’activation du plasminogène contenu dans le caillot par un tissu plasminogen activator (tPA) d’origine endothéliale.
La plasmine libère des produits de dégradation de la fibrine (PDF), comme le D-dimère, et exerce également une action régulatrice sur la coagulation en favorisant la dégradation des facteurs V, VII, IX et XI [15, 26].
Sans remettre en cause fondamentalement ces mécanismes, les concepts les plus récents reconsidèrent le déroulement de l’hémostase comme une suite de réactions localisées essentiellement à la surface des membranes cellulaires dont les phospholipides jouent un rôle essentiel de support à la formation des complexes enzymatiques (encadré 2).
• L’anticoagulant naturel le plus puissant est une enzyme circulante d’origine hépatique, l’antithrombine III qui assure 50 à 70 % de l’inhibition naturelle de la thrombine, ainsi que les facteurs IX, X, XI et XII de la cascade de coagulation [20]. Son cofacteur naturel, l’héparine, peut intensifier son activité jusqu’à 2 000 fois [7, 19, 20].
• Les principaux agents d’inhibition de la fibrinolyse et de stabilisation du caillot sont le plasminogen activator inhibitor (PAI) d’origines endothéliale et plaquettaire, qui inactive le tPA et l’a-antiplasmine qui exerce une inhibition compétitrice sur le plasminogène contenu dans le caillot [15, 19].
• Enfin, la protéine C, synthétisée par le foie et vitamine K-dépendante, a une action à la fois inhibitrice sur la coagulation, par inactivation des facteurs V et VIII, et promotrice sur la fibrinolyse, par interférence avec le PAI [15, 26].
• De plus, de multiples phénomènes d’autorégulation et d’auto-amplification interviennent entre les différents agents de l’hémostase et de la fibrinolyse [18, 19, 26]. Ainsi, la thrombine, agent final de la cascade de coagulation, exerce des effets activateurs en amont sur les facteurs V, VIII et XI, ainsi que sur l’agrégation plaquettaire. À l’inverse, les produits de dégradation de la fibrine, lorsqu’ils sont présents en grande quantité, peuvent avoir un effet inhibiteur sur l’agrégation plaquettaire, et la synthèse de thrombine et de fibrine [15].
• Ces nombreuses interactions et régulations réciproques font de l’hémostase un phénomène physiologique complexe en équilibre fragile. La moindre perturbation dans la production, la consommation ou l’activation des thrombocytes ou des facteurs plasmatiques de coagulation et de régulation est susceptible de déclencher des déséquilibres sévères entre la formation et l’élimination de la fibrine, avec à la clé un défaut d’hémostase primaire ou secondaire (saignements), ou, à l’inverse, une activation excessive de la coagulation (thromboses vasculaires, coagulation intravasculaire disséminée [CIVD]).
La fonction hémostatique primaire peut être altérée soit par une quantité insuffisante de plaquettes circulantes, soit par un défaut d’activation de ces dernières (tableau).
Une thrombocytopénie (inférieure à 100 x 109 plaquettes/l) résulte beaucoup plus souvent d’une consommation excessive, d’une perte massive ou d’une destruction immunitaire des plaquettes que d’un défaut de production [17, 19, 26].
• La consommation excessive de plaquettes circulantes est une des complications possibles d’un trouble de la coagulation, quelle qu’en soit la nature. Elle peut aussi survenir à la suite d’une vasculite étendue [19, 26]. C’est le cas, par exemple, lors de purpura hémorragique secondaire à certaines infections à streptocoques.
• Lors d’une hémorragie massive, une baisse de la numération plaquettaire peut survenir en même temps que l’anémie [26].
• Les thrombocytopénies par destruction périphérique à médiation immunitaire peuvent être primaires (idiopathique, néonatale), ou, beaucoup plus souvent, secondaires à une administration médicamenteuse (phénylbutazone, sulfamidés, pénicilline G), à une infection systémique (anémie infectieuse, ehrlichiose) ou encore à un phénomène néoplasique, en particulier le lymphosarcome [2, 17, 19, 26].
• Une thrombocytopénie par séquestration splénique peut aussi être observée lors de toute affection s’accompagnant de splénomégalie, qu’elle soit tumorale, infectieuse, parasitaire (babésiose) ou hémodynamique (insuffisance cardiaque, congestion passive à la suite d’un déplacement de côlon à gauche) [20, 26].
De rares cas de thrombocytopénie par défaut de production ont été décrits chez le cheval, associés à des maladies myéloprolifératives, à des intoxications (œstrogène, phénylbutazone, chloramphénicol) ou, très rarement, à des maladies auto-immunes avec une destruction de mégacaryocytes, précurseurs plaquettaires médullaires [20, 25-27]. Dans ces affections, une anémie et/ou une pancytopénie sont généralement présentes et dominent le tableau biologique.
Des déficiences de l’hémostase primaire peuvent occasionnellement être observées sans anomalie de la numération plaquettaire. La présence de pétéchies et l’allongement du temps de saignement constituent alors les principaux signes d’appel :
– une thrombasthénie héréditaire, dite “maladie de Glanzmann”, est occasionnellement observée chez le cheval [10, 14]. Il s’agit d’une mutation génétique affectant les récepteurs plaquettaires du fibrinogène, qui empêche l’activation des thrombocytes et la stabilisation du clou plaquettaire ;
– une déficience du facteur de von Willebrand, nécessaire à l’adhésion plaquettaire sur le collagène sous-endothélial vasculaire a aussi été décrite dans les races quarter horse et pur-sang [1, 13, 23]. Plusieurs types de maladie de von Willebrand sont rapportés : il peut s’agir d’un déficit quantitatif en facteur de von Willebrand ou d’une anomalie qualitative, parfois acquise, le rendant non fonctionnel [23].
Certaines formes de maladie de von Willebrand sont associées à un déficit en facteur VIII de la coagulation. Une perturbation de l’hémostase secondaire est alors associée aux anomalies de la fonction plaquettaire [1].
Les perturbations de l’hémostase secondaire se manifestent aussi bien par défaut que par excès et ont des origines très diverses : septiques, traumatiques, métaboliques, toxiques ou héréditaires.
Une activation massive, non régulée, de la coagulation sanguine et de la fibrinolyse, sans rapport avec une lésion vasculaire ni localisation précise, caractérise la coagulation intravasculaire disséminée.
Chez le cheval, l’activation de la coagulation par les endotoxines circulantes est la principale cause de cette affection grave qui n’apparaît pas de façon isolée, mais généralement en complication d’une affection gastro-intestinale sévère, d’une chirurgie digestive, d’un sepsis systémique ou d’un état de choc avancé [4, 6, 9, 22, 28].
Parfois, un état de CIVD chronique et d’expression plus discrète s’installe au cours de l’évolution d’une affection rénale ou digestive chronique, d’un syndrome paranéoplasique ou hémolytique [5, 19, 26].
Les endotoxines bactériennes sont capables de stimuler directement le facteur XII, mais l’initiation de la CIVD se fait en majeure partie par l’activation des macrophages qui libèrent des cytokines à activité procoagulante (tumor necrosis factor, prostaglandines) et pro-agrégante (platelet activating factor) (figure 3) [5, 15, 19].
La libération massive et persistante de thrombine activée dépasse les capacités de régulation de l’antithrombine III, dont les réserves s’épuisent, et se solde par la formation de multiples microthrombus responsables de nécrose ischémique rénale, digestive ou encore digitale.
Pendant cette phase, qui peut aussi bien rester transitoire et subclinique que s’accompagner de signes généraux sévères, le cheval est prédisposé à développer une thrombose des gros vaisseaux lors de la moindre effraction pariétale, même une simple prise de sang [6, 15, 19, 28].
À terme, l’épuisement des facteurs de coagulation circulants et des plaquettes, associé au rétrocontrôle négatif exercé sur la coagulation par les produits de dégradation de la fibrine, entraîne une déficience de l’hémostase à la fois primaire et secondaire.
Cette dernière étape, marquée par une hypocoagulabilité sanguine générale et l’apparition de saignements disséminés et incoercibles, est très souvent fatale [5, 19].
Les intoxications aux coumariniques entraînent une inhibition de la synthèse hépatique des facteurs de coagulation vitamine K-dépendants (II, VII, IX, X). La demi-vie du facteur VII étant la plus brève, la voie extrinsèque de la coagulation est affectée la première [5, 20].
La fraction circulante libre des coumariniques étant responsable de leur toxicité, cette dernière est accentuée par une hypoprotéinémie ou la présence de substances se fixant de façon compétitive sur les protéines plasmatiques, comme les anti-inflammatoires non stéroïdiens [5].
En médecine équine, les anticoagulants coumariniques ne sont plus utilisés comme principes thérapeutiques et les intoxications sont essentiellement d’origine alimentaire : ingestion directe de céréales traitée aux rodenticides à forte concentration ou consommation de coumariniques “naturels” dans des fourrages fermentés [5, 16, 19]. En raison des doses toxiques nécessaires assez importantes et de la synthèse naturelle de vitamine K dans le côlon, ces intoxications restent rares chez le cheval.
L’absorption digestive de la vitamine K étant conditionnée par la présence de bile, une insuffisance hépatique et/ou une cholestase sévère, qu’elles soient aiguës ou chroniques, ont des conséquences semblables à celles d’une intoxication aux coumariniques, à savoir une déplétion progressive des facteurs de coagulation vitamine K-dépendants [5, 19, 20].
Si, de façon paradoxale, la concentration circulante d’antithrombine III est généralement beaucoup moins affectée par l’insuffisance hépatique, elle est néanmoins réduite par des pertes protéiques massives et chroniques. Une entéropathie exsudative ou un syndrome néphrotique peuvent ainsi favoriser l’installation insidieuse d’un état d’hypercoagulabilité et une prédisposition aux thromboses vasculaires, sans activation massive de la coagulation comme dans la CIVD, mais par déficience de l’activité anticoagulante plasmatique naturelle [20].
De rares affections héréditaires peuvent affecter la synthèse des facteurs VIII, IX, XI et prékallicréine, et altèrent spécifiquement le déroulement de la voie intrinsèque de la coagulation.
L’hémophilie A, déficience héréditaire en facteur VIII, est décrite dans les races pur-sang, trotteur et quarter horse [5, 19]. Sa transmission sur le mode autosomal récessif est liée au chromosome X et atteint essentiellement les mâles.
Les individus touchés manifestent généralement des signes cliniques assez sévères et leur espérance de vie est très réduite.
La déficience en prékallicréine a été décrite dans quelques lignées de chevaux belges et de chevaux miniatures [11]. Son expression clinique est généralement très fruste, probablement en raison du rôle amplificateur mais non déterminant de la prékallicréine dans le déroulement de la voie intrinsèque de la coagulation.
L’hémostase est une fonction physiologique complexe constamment sollicitée, même en dehors de tout contexte pathologique, et dont l’équilibre peut être perturbé par une grande variété d’affections, pour la plupart inflammatoires, traumatiques ou septiques. De plus, tout déficit fonctionnel d’une étape de l’hémostase est susceptible d’entraîner secondairement un épuisement de tous ses autres agents cellulaires et biochimiques par activation excessive et surconsommation. Les maladies primaires de la fonction hémostatique sont beaucoup plus rares dans l’espèce équine, et, de ce fait, moins connues et moins recherchées. Certaines affections héréditaires requièrent cependant l’attention, ne serait-ce que pour leur prophylaxie génétique.
Enfin, des déficits même discrets de l’hémostase primaire sont parfois les signes les plus précoces d’un syndrome paranéoplasique ou d’un processus infectieux chronique qui peut avoir une importance médicale et sanitaire non négligeable.
→ Les artefacts sont nombreux lors de la mesure de la numération plaquettaire chez le cheval. La réitération du dosage sur tube citraté et/ou la validation de la numération plaquettaire sur frottis sanguin sont nécessaires avant de poursuivre les examens complémentaires.
→ Des affections à médiation immune sont la plupart du temps en cause lors de thrombocytopénie avérée sans altération de l’état général.
→ La coagulation intravasculaire disséminée est le trouble le plus fréquent et le plus grave de la fonction hémostatique. Elle apparaît la plupart du temps sous forme aiguë au cours de l’évolution d’une affection métabolique ou digestive sévère dont elle aggrave le pronostic, mais peut aussi exister sous une forme subclinique plus difficile à détecter.
→ Les thrombocytes sont les plus petites cellules sanguines. Ce sont des éléments cytoplasmiques anucléés de 1 à 4 µm de diamètre, de coloration acidophile, de forme sphérique, discoïde ou allongée, qui contiennent essentiellement des granules de sécrétions, des microfilaments contractiles, et un complexe de canalicules internes issus de l’invagination de la membrane lipidique externe [16, 22]. Les plaquettes sont issues de la fragmentation cytoplasmique de leurs précurseurs médullaires, les mégacaryocytes, et leur durée de vie dans la circulation sanguine est de l’ordre de 4 à 5 jours, avant destruction par les macrophages, principalement spléniques, ou par consommation au cours de l’hémostase [22].
→ La numération plaquettaire est normalement supérieure à 100 x 109/l chez le cheval, mais les conditions de réalisation et de conservation des prélèvements sont à prendre en considération avant d’interpréter les résultats : une altération des plaquettes conduit facilement à une thrombocytopénie par artefact sur un échantillon sanguin conservé plus de 12 heures ou soumis à des variations de température importantes, ou lorsque la ponction veineuse a été difficile et que des microparticules tissulaires ont été aspirées [22, 24, 26]. Si un résultat anormalement bas est obtenu, il doit, de préférence, être confirmé par plusieurs dosages répétés, dont au moins un sur tube citrate. En effet l’EDTA provoque parfois une agrégation plaquettaire qui fausse la numération, surtout sur les compteurs automatiques [24]. Une thrombocytopénie évaluée sur automate devrait toujours in fine être confirmée sur frottis sanguin.
→ La membrane des plaquettes est recouverte d’une couche de glycoprotéines qui empêche leur adhésion à l’endothélium vasculaire sain, mais en revanche favorise leur adhésion sur le collagène sous-endothélial exposé lors de brèche vasculaire, par l’intermédiaire d’une glycoprotéine endothéliale, le facteur de von Willebrand qui forme un complexe avec le facteur VIII de la coagulation [16, 26].
→ L’agrégation qui fait suite à cette adhésion est caractérisée par une modification morphologique très importante des plaquettes avec hypertrophie, contraction des microfilaments cytoplasmiques, et formation de pseudopodes [16]. Cette agrégation d’abord réversible nécessite le contact des plaquettes avec des molécules de fibrinogène et des ions calcium.
→ Initiée par des facteurs endothéliaux comme le platelet activating factor, l’activation plaquettaire débute par la dégranulation d’adénosine diphosphate (ADP), adénosine triphosphate (ATP), sérotonine, facteurs plaquettaires 3 et 4, et est entretenue par la synthèse de thromboxane A2 à partir de l’acide arachidonique contenu dans les membranes plaquettaires et métabolisé par la voie de la cyclo-oxygénase. L’inhibition de cette dernière est à l’origine de l’effet antiagrégant des anti-inflammatoires non stéroïdiens. L’activation rend l’agrégation irréversible et permet la formation du clou plaquettaire définitif. Ce clou n’est pas suffisant en lui-même pour stopper l’hémorragie sur une brèche vasculaire de grande taille, mais joue un rôle essentiel pour colmater les milliers de microlésions endothéliales qui se forment chaque jour dans les petits vaisseaux [16, 22]. L’un des signes cliniques les plus révélateurs de thrombocytopénie est la formation de micro-hématomes sous-endothéliaux appelés pétéchies (photos 1 et 2) [16, 22, 26].
→ Le déroulement de l’hémostase est désormais reconsidéré comme une suite de formations de complexes impliquant les phospholipides des membranes cellulaires et divisées en trois phases localisées sur deux surfaces cellulaires : surface des cellules sous-endothéliales exposées par la brèche vasculaire et celle des plaquettes.
→ La phase d’initiation à la surface des cellules sous-endothéliales débute lors de l’extravasation du plasma, et l’activation du facteur VII par le facteur tissulaire, aboutissant par l’intermédiaire des facteurs IX, X et V à la formation d’une petite quantité de thrombine sur la surface cellulaire.
→ La phase d’amplification correspond à l’adhésion et à l’activation des thrombocytes, et la formation d’un complexe entre facteurs V, VIII et phospholipides sur la membrane plaquettaire.
→ La phase de propagation à la surface du clou plaquettaire aboutit par la voie commune de la coagulation à la libération d’une quantité plus importante de thrombine et à la formation du caillot de fibrine.
Aucun.
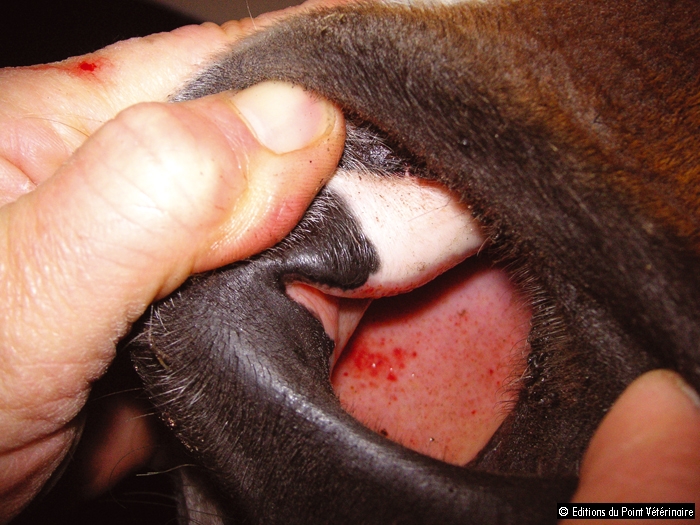
Photos 1. Pétéchies sur la muqueuse du septum nasal (1) et la conjonctive palpébrale (2) d’un cheval atteint de thrombocytopénie auto-immune.
Cliché : V. Deniau.
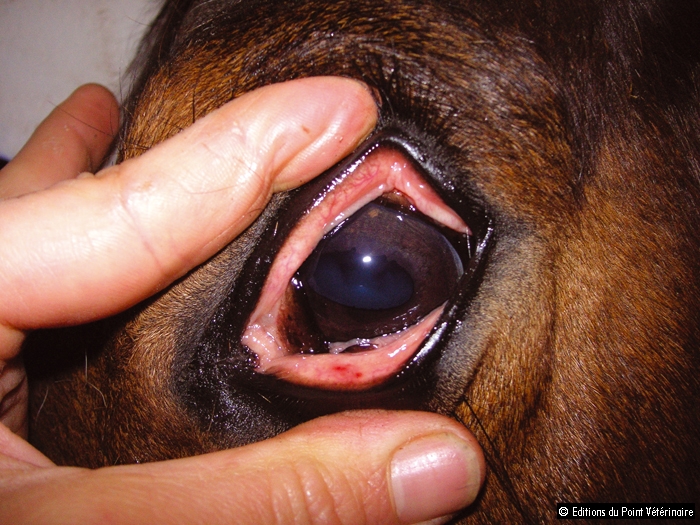
Photos 2. Pétéchies sur la muqueuse du septum nasal (1) et la conjonctive palpébrale (2) d’un cheval atteint de thrombocytopénie auto-immune.
Cliché : V. Deniau.
Figure 2 : Résumé des principaux mécanismes de la coagulation et de la fibrinolyse, et sites d’action des agents de régulation



